
Softwares

ADRP-ML-NMF
Identifying and controlling adverse drug reactions is a complex problem in the pharmacological field. Despite the studies done in different laboratory stages, some adverse drug reactions are recognized after being released, such as Rosiglitazone. Due to such experiences, pharmacists are now more interested in using computational methods to predict adverse drug reactions.
Software Page
DRP-VEM
Traditional drug discovery methods are costly and time-consuming. The use of the existing approved drugs for treating another disease is called drug repositioning, a common strategy to overcome traditional drug discovery issues. Current drug repositioning approaches are vastly applied to machine learning methods. The performance of these methods seriously depends on types of features, their representations and the training dataset. Most drug repositioning approaches integrate different features and varied representations. As they do not consider the redundancy of features, it remains a challenge in facing drug repositioning problem.
Software Page
TranDTA
TranDTA is the first method that applies transformers to extract feature of protein sequence and uses transformer representations in drug target binding affinity (DTBA) prediction. This model only uses inputs as 1D representations. It can also be run without limitations on resources (memory, CPU and GPU) due to the use of pretrained representations for inputs. This model can be helpful in drug development process.
Software Page
CNDP_PPI_networks
The data set used in the paper contains the PPI networks of E. coli and S. cerevisiae which are extracted from the Database of Interacting Proteins (DIP). To label the proteins as essential/non-essential, the essential genes data of these species are collected from DEG database.
Software Page
WCOACH
WCOACH is a graph clustering algorithm to detect protein complexes in weighted Protein-Protein Interaction (PPI) networks. protein complexe prediction is a chalanging problem in computational biology. WCOACH algorithm is a modification of COACH method to solve this problem in weighted PPI networks.
Software Page
RNASHAPE
RNA molecules play important and fundamental roles in biological processes. Frequently, the functional form of single-stranded RNA molecules requires a specific tertiary structure. Classically, RNA structure determination has mostly been accomplished by X-Ray crystallography or Nuclear Magnetic Resonance approaches. These experimental methods are time consuming and expensive. In the past two decades, some computational methods and algorithms have been developed for RNA secondary structure prediction. In these algorithms, minimum free energy is known as the best criterion. However, the results of algorithms show that minimum free energy is not a sufficient criterion to predict RNA secondary structure. These algorithms need some additional knowledge about the structure, which has to be added in the methods. Recently, the information obtained from some experimental data, called SHAPE, can greatly improve the consistency between the native and predicted RNA secondary structure.
Software Page
ESD-Fold
ESD-Fold is a new method to predict RNA secondary structure based on both minimum free energy and SHAPE- directed pseudo-free energy. For each RNA sequence, this method employs a base pair probability matrix of all potential stems to construct a population of RNA secondary structures. It should be noted that RNAfold software, that is free of charge for academic users and available in address "http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi", is applied to compute the matrix. Then, for each constructed secondary structure, its SHAPE data is simulated. After that, an evolutionary algorithm is used to improve each structure based on both free and pseudo-free energies. Population generation is continued until the best individual in the two last populations remains fixed. Then, a structure with minimum summation of free energy and pseudo-free energy is considered as the predicted RNA secondary structure.
Software Page
GRNAs
The GRNAs predicts RNA-RNA interaction structures including kissing hairpins. GRNAs computes the minimum free energy (MFE) structures by using a novel genetic algorithm
Software Page
GAPSSIF
GAPSSIF is a novel genetic algorithm which uses native secondary sub-structures is proposed to solve Protein Secondary Structure Inverse Folding (PSSIF) problem. In essence, evolutionary information can lead the algorithm to design appropriate amino acid sequences respective to the target secondary structures. Furthermore, they can be folded to tertiary structures almost similar to their reference 3D structures.
Software Page
HRDSSD
HRDSSD is a new method for accurate design of RNA sequences based on their secondary structures using SHAPE data as pseudo-free energy. We first use a stochastic model to simulate SHAPE data based on a set of 16S and 23S ribosomal RNA sequences. Then, a harmony search algorithm is applied to accurately predict RNA sequence using free energy and simulated SHAPE data.
Software Page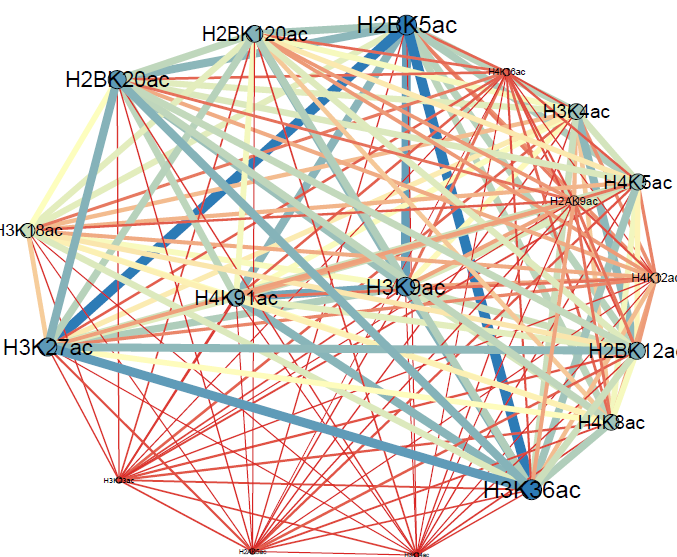
HAmTFBS-Net
Code provided for "The Assessment of Histone Acetylation Marks in the Vicinity of Transcription Factor Binding Sites in Human CD4+T Cell Using Information Theory Methods" article
Software Page
CEBCP
Sometimes, drugs are withdrawn due to low bioactivity that is not observed during the experimental procedure. Thus, predicting bioactivity classes during the lead optimization reduces the failures risk and improves the enhancement of compound bioactivities. Recent studies demon-strate a relationship between the chemical structure of compounds and their bioactivity, i.e., struc-ture-activity relationship (SAR), without considering the complex relationship between drugs and bioactivity classes.
Software Page